
The Consortium for Computational Physics and Chemistry (CCPC) researches catalysts at atomic scale to enable innovative technologies for cost-effective and energy-efficient biomass-to-fuel processes.
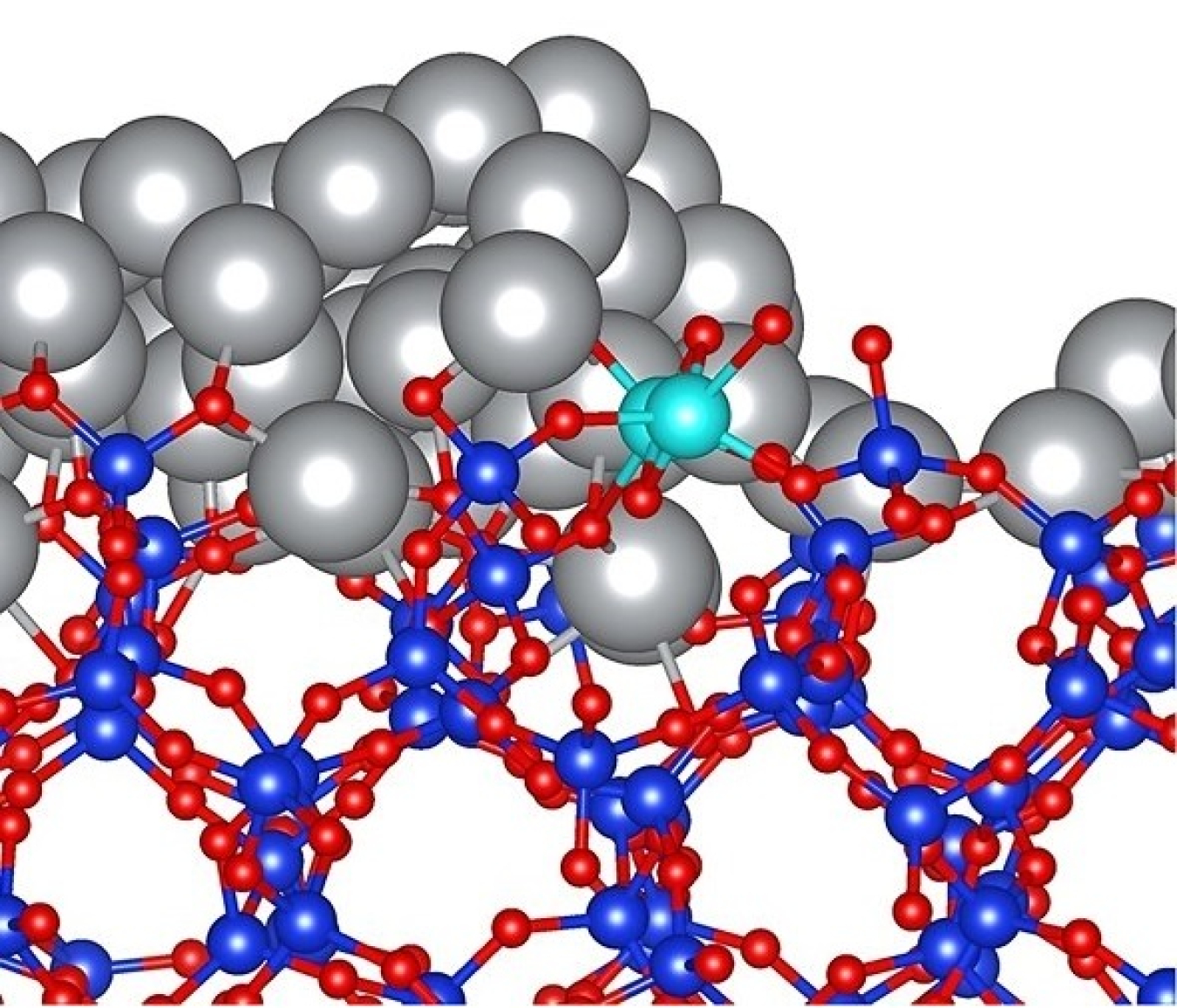
An image of a catalyst model at atomic scale used by CCPC to study the rate of conversion. Image courtesy of Roger Rousseau, Pacific Northwest National Laboratory.
There are critical chemical reactions that occur on the surface of catalysts in order to transform lignocellulosic biomass into biofuels and other bioproducts.
A Primer on Biomass-to-Fuel Processes
In the first stages of biomass-to-fuel processes, deconstruction and fractionation of biomass into chemicals such as oils, alcohols, sugars, and gases occur. Then, the resulting chemicals are catalytically converted into intermediate chemistries, fuels, and co-products.
The catalytic conversion occurs at atomic and molecular scales when the gas or liquid reactant species adsorbs on the surface of a solid catalyst. The chemical and electronic structure of the catalyst surface and the thermodynamic energy states of the reactant molecule dictate the rate of chemical conversion as well as the resulting product chemistry.
Overall, the objective of biomass-to-fuel process catalyst design is to enable low energy (low temperature) chemical conversion to select chemicals suitable for blending into transportation fuels through an atomic scale understanding of reaction mechanisms and structure/property relationships. Research at CCPC also focuses on cost-effective materials to enable energy efficient catalytic conversion technologies that are economically feasible.
Atomic-Scale Computational Tools in the CCPC
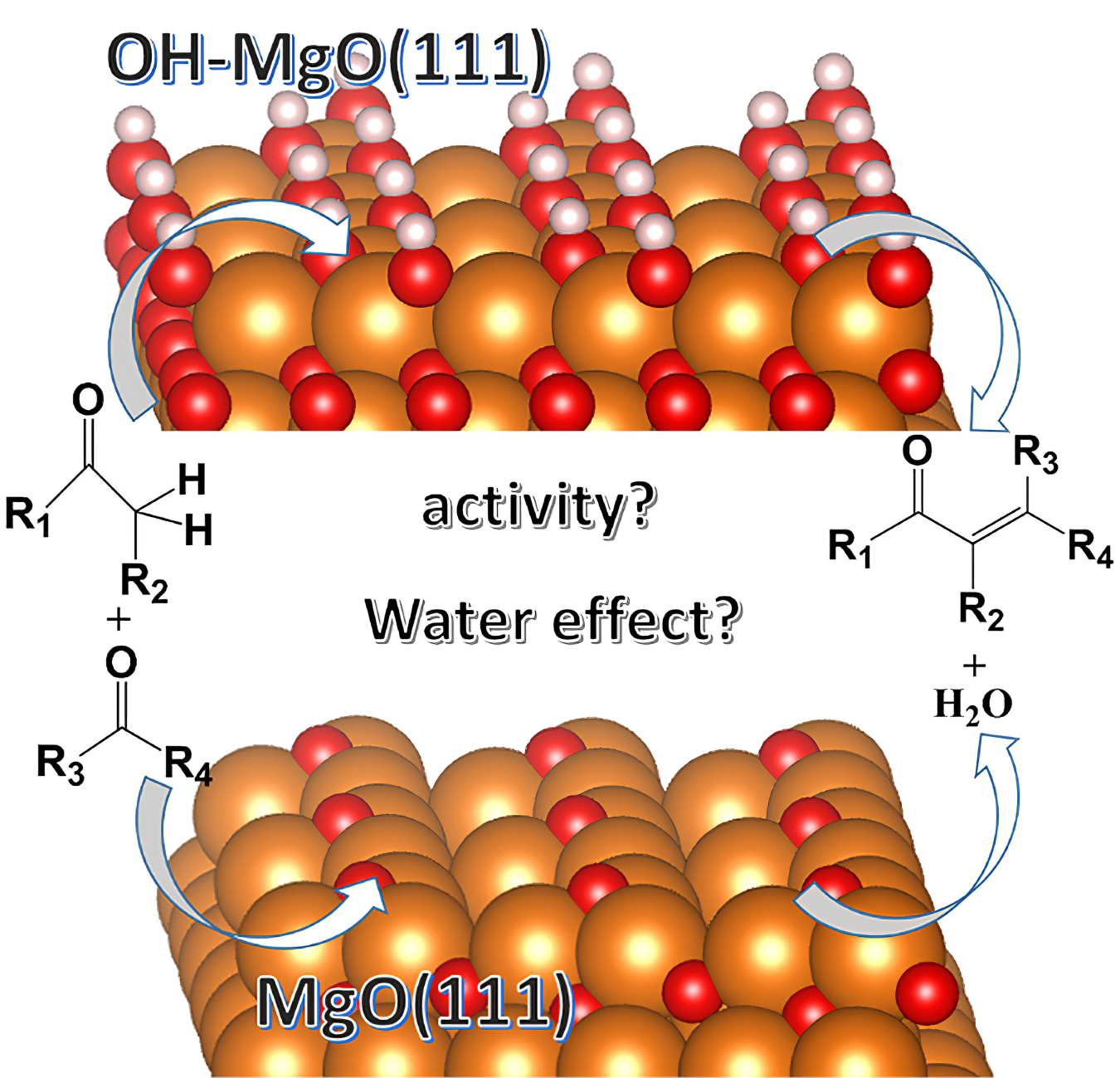
Figure 1. Density Functional Theory model of a MgO(111) catalyst surface with (top) and without (bottom) water adsorbed on the catalyst surface. The model can be used to show the effects of adsorbed water on the difference in activity for specific hydrocarbons reacting on the catalyst surface. Image courtesy of Rajeev, Assary, Argonne National Laboratory.
An important computational tool for understanding catalytic conversion processes is Density Functional Theory (DFT). DFT is based on the quantum mechanical theory of atomic electronic structure. By modeling a catalyst surface with DFT, the binding energy of adsorbed reactants can be determined, which gives important information on the preferred chemical reactions.
Since DFT computations are dependent on catalyst structure and reactant chemistry, multiple DFT calculations are typically required to understand the range of chemical conversion steps to a specific fuel chemical.
The CCPC’s DFT models enable measurement of the critical energy transitions that occur when bioenergy molecules interact with the catalytic surface at atomic scales.
DFT models also help understand the impact of competitive or synergistic molecular compounds and impurities that adsorb on the catalyst surface in real biomass-to-fuel process streams. An example of this would be that water adsorption on a catalyst can impact catalyst activity for hydrocarbon conversion (Figure 1).
Ab Initio Molecular Dynamics Tool
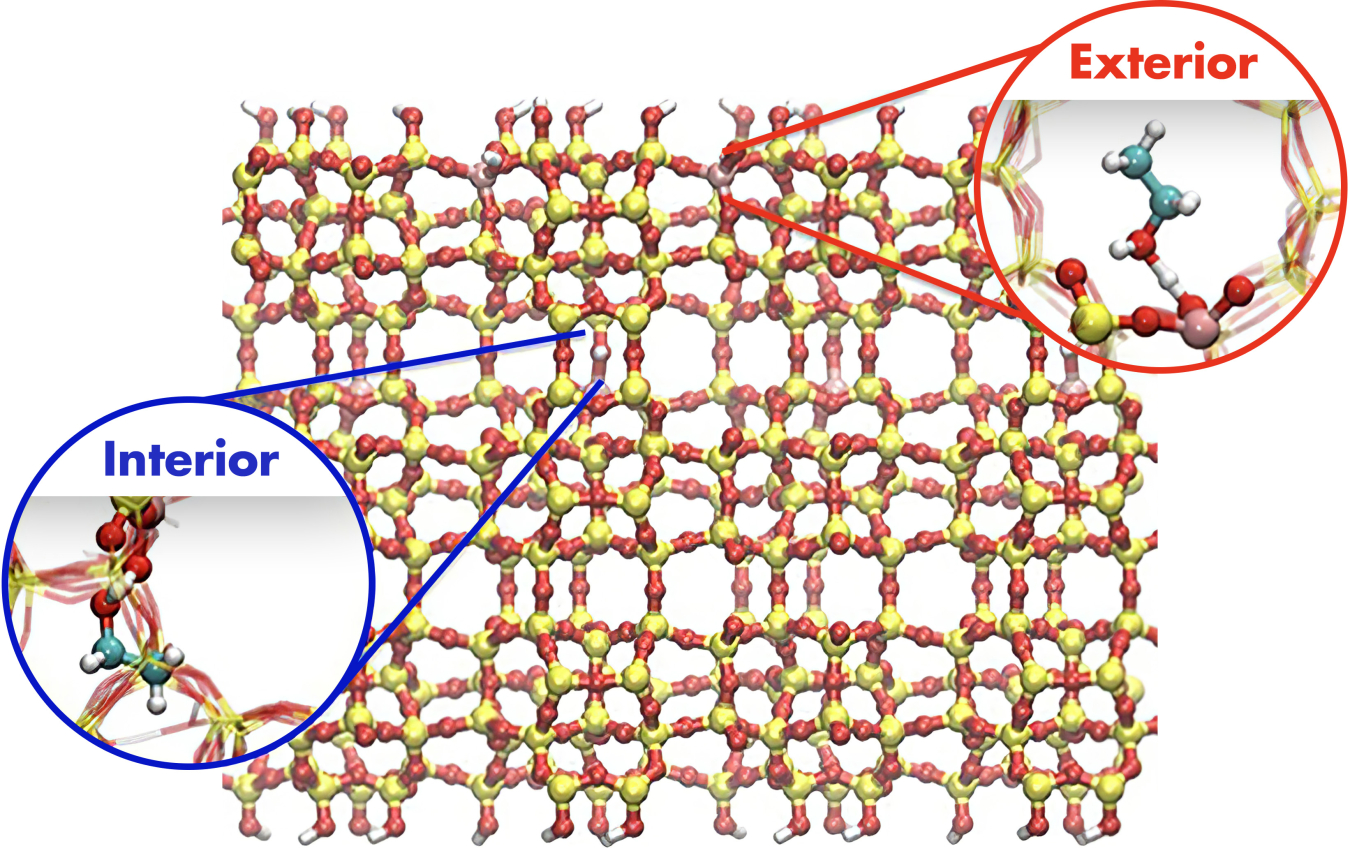
Figure 2. Model representation of the molecular-scale structure of a zeolite material commonly used in biomass-to-fuel conversion processes with insets showing hydrocarbon bonding to the interior and exterior parts of the zeolite structure. Image courtesy of Roger Rousseau, Pacific Northwest National Laboratory.
Another tool utilized in CCPC atomic scale tasks is ab initio molecular dynamics (AiMD). This tool combines electronic structure (e.g., DFT) and statistical mechanics to compute thermodynamics and dynamics at elevated temperatures. Many catalysts used for bioenergy conversion have inherent porosity over wide ranging scales.
For example, a specific class of materials known as zeolites incorporates very small pores of 0.1 nm to 1 nm similar to the size of individual molecules. These molecular-sized pores confine reactants changing their free energy in ways that enable zeolites to achieve improved conversion efficiencies for selective chemical reactions.
AiMD enables the modeling of the molecular vibrations and confined molecular motions that can be used to predict the reactivity of these small pores. Such understanding provides guidance on selecting the right zeolite material for a given bioenergy conversion reaction.